Startups love prototypes. They’re tangible, testable, and exciting. But if you want to commercialize a Class II or Class III medical device, the physical product is only half the story. The other half? Telling the FDA why the device is safe and effective.
That story is captured in your Design History File (DHF), the structured documentation of your design controls required by 21 CFR 820.30. For first time startups, this may be unfamiliar territory. At Medical Murray, we help you align technical development with regulatory expectations from the start, so your device is ready for submission, transfer, or scale-up.
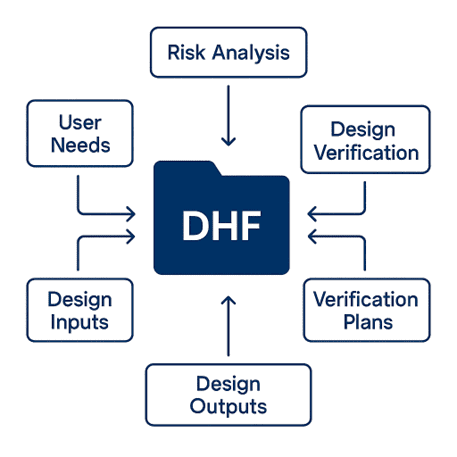
What’s in a Design History File And Why It Matters
Your DHF documents the evolution of the device from concept to design freeze. It connects user needs to design inputs, risk analysis, verification plans, and design outputs, all in a way that supports regulatory and quality compliance.
A well-built DHF is essential if you plan to:
- Submit a 510(k), De Novo, or PMA
- Transfer to a contract manufacturer
- Attract quality-focused investors or partners
- Demonstrate compliance with ISO 13485 or FDA QSR
You don’t need a full regulatory team to get started, but you do need a plan.
Roadmap: From Concept to Medical Device DHF
Phase 1: Concept Development
Aligning User Needs with Design Inputs
We start with the why. Understanding clinical context, user personas, intended use, and functional expectations lays the foundation for traceability to design inputs. We help translate insights into User Needs and Engineering Requirements. We define early design inputs that are testable, measurable, and linked to the problem you’re solving. You’ll walk away with a Design and Development Plan, a preliminary BOM, and early documentation aligned with FDA expectations.
Phase 2: Feasibility Builds
Verifying Against Early Requirements
With design inputs defined, we build and test early prototypes to support feasibility and de-risk development. We establish risk-based plans using the ISO 14971 framework, including Hazard Analyses and FMEAs. Our units are built with traceable lot history and tested using custom, documented test methods.
Phase 3: Pre-Design Verification
Preparing for Design Freeze
We optimize your design and process for formal verification. This includes generating detailed design outputs (drawings, specs, BOMs), refining manufacturing instructions, and drafting Design Verification Test Plans and protocols. Trace matrices ensure each input links to a corresponding output.
These builds support bench testing and simulated use testing, and are structured for seamless transition into controlled DV builds. All results feed into your DHF through structured Design Reviews.
Phase 4: Design Verification
Regulatory Ready
We lock your Design Master Record (DMR) and execute DV builds under controlled conditions. We manage test execution including bench testing, biocompatibility, shelf-life, and distribution simulation. We provide protocols, raw data, and formal reports for your regulatory submission.
We also finalize the Risk Management File and complete traceability and justifications for any deviations. This acts as your proof of performance; that you are ready for inspection, audit, or due diligence.

How Medical Murray Helps
We support Class II and Class III medical device developers with:
- Design inputs built around real clinical needs
- Prototypes aligned with verification and design freeze goals
- DHF documentation ready for submission, transfer, or scale-up
- Processes that meet ISO 13485 and FDA QSR standards
We bridge engineering and regulatory from day one so you’re not scrambling to retrofit documentation at the last minute.
Steps are documented. Deliverables feed your DHF. From optional protocols to full validation packages, our medical device design services balance speed, quality, and compliance.
For Startups: Start with Strategy
- Document early and often; even rough notes are better than nothing
- Maintain traceability: user needs → inputs → outputs → verification
- Plan for submission, not just the prototype
- Work with a partner who understands both engineering and FDA expectations
Whether you’re pursuing a 510(k), IDE, or CE Mark, we help you establish a regulatory-ready foundation from the start, so you’re not reinventing the wheel later. Contact us to discuss how we can support your project in becoming regulatory-ready.
Looking for an expert partner to develop your medical device concept? Contact Medical Murray today!